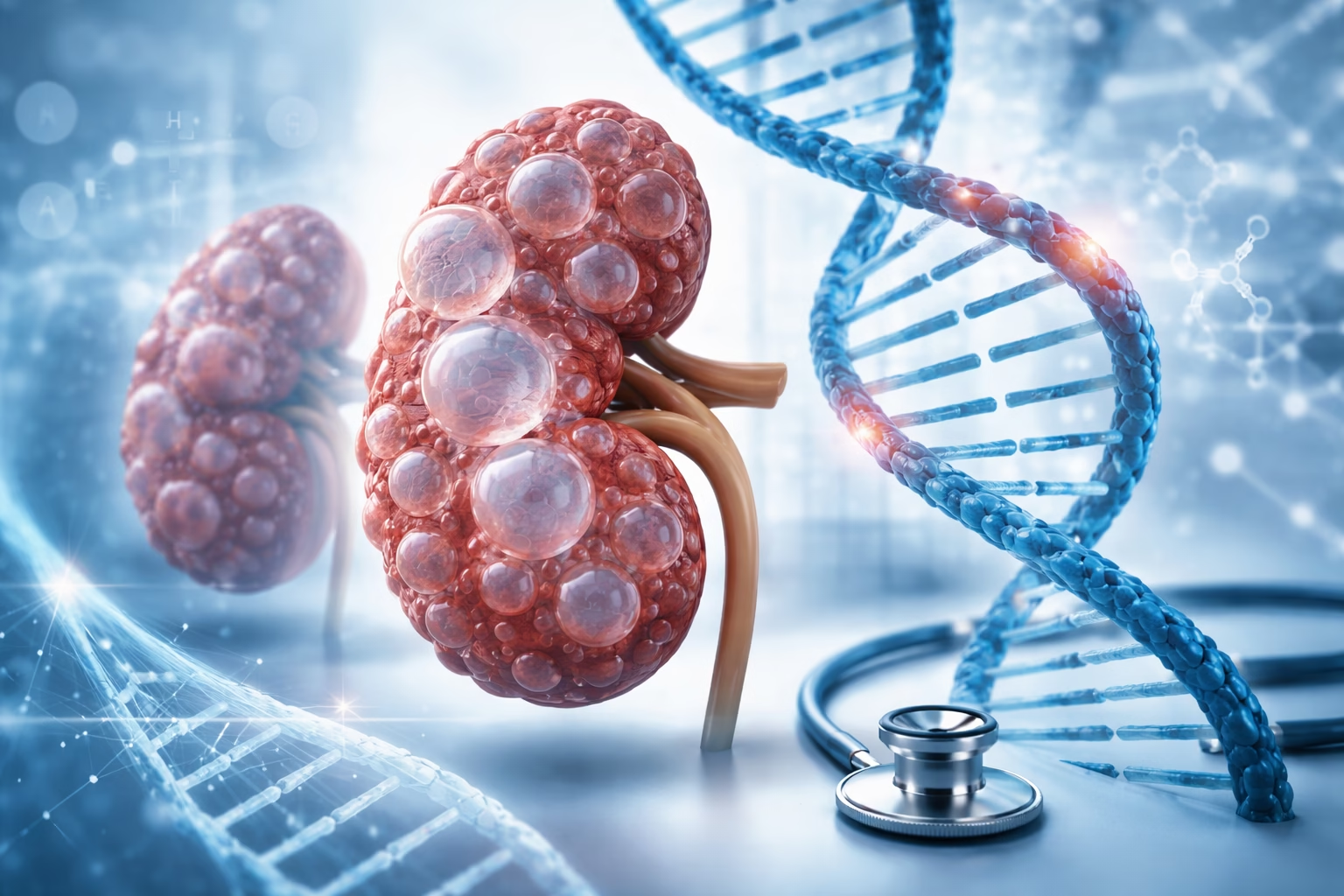
Polycystic Kidney Disease (PKD) is a hereditary disorder characterized by the progressive development of fluid filled cysts within the kidneys. Over time, these cysts enlarge and multiply, causing the kidneys to increase in size and gradually lose their ability to function properly. PKD is considered one of the most common inherited kidney diseases and affects millions of individuals worldwide. The condition can significantly impair renal function and may eventually lead to chronic kidney disease or kidney failure if not properly managed.
- Mutations in PKD1 and PKD2 disrupt kidney cell signaling; PKD1 typically causes earlier, more aggressive progression.
- ADPKD usually manifests in adulthood; ARPKD is less common and severe, often appearing in infancy or early childhood.
- Imaging like ultrasound, CT, and MRI plus genetic testing and lab studies enable early detection and disease monitoring.
- Control blood pressure, consider vasopressin receptor antagonists to slow cyst growth, manage pain, lifestyle changes, dialysis or transplant if needed.
Understanding the genetic origins of PKD and the available treatment strategies is essential for early diagnosis, disease monitoring, and improved long term outcomes. Although there is currently no definitive cure for the disease, advances in medical management and supportive therapies have significantly improved the quality of life for individuals living with PKD.
Understanding Polycystic Kidney Disease
Polycystic Kidney Disease involves the formation of numerous cysts within kidney tissue. These cysts are small fluid filled sacs that gradually expand over time. As they enlarge, they compress surrounding kidney structures, disrupt normal filtration processes, and ultimately reduce kidney efficiency.
In healthy kidneys, blood is filtered to remove waste products and maintain fluid and electrolyte balance. However, in individuals with PKD, the presence of cysts interferes with this filtration process. The kidneys may become significantly enlarged, sometimes several times their normal size.
PKD is generally categorized into two main types based on inheritance patterns and the age at which symptoms typically appear.
Related Posts:




Autosomal Dominant Polycystic Kidney Disease (ADPKD)
Autosomal Dominant PKD is the most common form of the disease. Symptoms usually develop during adulthood, typically between the ages of 30 and 50. Individuals who inherit a single mutated gene from one parent can develop the condition.
ADPKD progresses gradually and may remain undetected for many years until complications arise or imaging tests reveal enlarged kidneys.
Autosomal Recessive Polycystic Kidney Disease (ARPKD)
Autosomal Recessive PKD is a rarer and more severe form that often manifests in infancy or early childhood. In this type, a child must inherit defective genes from both parents in order to develop the disease.
ARPKD can cause significant kidney and liver complications early in life and often requires specialized medical care.
Genetic Causes of PKD
Polycystic Kidney Disease arises from mutations in specific genes responsible for regulating kidney cell growth and fluid balance. These genetic abnormalities lead to abnormal cellular signaling, resulting in the formation of cysts within the kidney tubules.
PKD1 Gene Mutation
The majority of PKD cases are associated with mutations in the PKD1 gene. This gene encodes a protein known as polycystin 1, which plays a crucial role in maintaining the structural integrity of kidney cells.
When mutations occur in the PKD1 gene, the signaling pathways that regulate cell growth become disrupted. Consequently, cells begin to proliferate abnormally, leading to cyst formation.
Individuals with PKD1 mutations often experience a more aggressive disease progression and may develop kidney failure earlier in life.
PKD2 Gene Mutation
Mutations in the PKD2 gene account for a smaller proportion of PKD cases. This gene produces polycystin 2, a protein involved in calcium signaling within kidney cells.
Although PKD2 mutations also lead to cyst formation, the disease progression is generally slower compared to PKD1 related PKD.
Genetic Transmission
PKD is typically inherited through a dominant pattern. This means that if one parent carries the defective gene, there is a fifty percent chance that the child will inherit the condition.
However, spontaneous genetic mutations can also occur in individuals with no family history of PKD.
Common Symptoms of PKD
The symptoms of Polycystic Kidney Disease vary depending on the stage and severity of the condition. In early stages, many individuals remain asymptomatic. As cysts grow larger, noticeable symptoms may begin to develop.
Common symptoms include:
• Persistent back or side pain
• High blood pressure
• Frequent urinary tract infections
• Blood in the urine
• Kidney stones
• Abdominal fullness due to enlarged kidneys
• Gradual decline in kidney function
In advanced cases, patients may develop complications such as chronic kidney disease, cardiovascular issues, or liver cysts.
Complications Associated with PKD
PKD can affect several organs beyond the kidneys. Because the disease involves systemic genetic abnormalities, other complications may arise.
Hypertension
High blood pressure is one of the earliest and most common complications of PKD. Enlarged kidneys can disrupt blood flow and activate hormonal pathways that increase blood pressure.
Kidney Failure
As cysts continue to grow, kidney tissue becomes progressively damaged. Eventually, the kidneys may lose their ability to filter waste products, leading to end stage renal disease.
Liver Cysts
Many individuals with PKD also develop cysts in the liver. Although these cysts rarely impair liver function, they can cause discomfort or abdominal swelling.
Cardiovascular Complications
PKD has been associated with abnormalities in blood vessels and heart valves. These complications may increase the risk of aneurysms or cardiovascular disease.
Diagnosis of Polycystic Kidney Disease
Early detection of PKD is crucial for slowing disease progression and preventing complications. Physicians typically use several diagnostic methods to identify the condition.
Imaging Tests
Ultrasound is the most common diagnostic tool used to detect kidney cysts. It allows physicians to visualize enlarged kidneys and determine the number and size of cysts present.
Other imaging methods may include:
• CT scans
• Magnetic resonance imaging (MRI)
These advanced techniques provide more detailed images and help monitor disease progression.
Genetic Testing
Genetic testing can identify mutations in the PKD1 or PKD2 genes. This method is particularly useful for individuals with a family history of PKD or those considering family planning.
Blood and Urine Tests
Laboratory tests may be used to evaluate kidney function. These tests measure indicators such as creatinine levels, electrolyte balance, and protein in the urine.
Treatment Approaches for PKD
Although there is currently no cure for Polycystic Kidney Disease, treatment focuses on slowing cyst growth, controlling symptoms, and preventing complications.
Blood Pressure Management
Controlling blood pressure is a critical component of PKD treatment. Medications such as ACE inhibitors or angiotensin receptor blockers are commonly prescribed to reduce strain on the kidneys.
Medications to Slow Disease Progression
Certain medications may help slow the development of kidney cysts. For example, vasopressin receptor antagonists have shown promise in reducing cyst growth in patients with ADPKD.
Pain Management
Chronic pain caused by enlarged kidneys may require medical management. Physicians may recommend analgesic medications or minimally invasive procedures to relieve discomfort.
Lifestyle and Dietary Modifications
Lifestyle changes play a significant role in managing PKD.
Recommended strategies include:
• Maintaining adequate hydration
• Limiting sodium intake
• Managing blood pressure through diet and exercise
• Avoiding smoking
• Reducing caffeine consumption
These lifestyle modifications can help support kidney health and reduce disease complications.
Dialysis and Kidney Transplant
In advanced stages of PKD, when kidney function declines significantly, renal replacement therapy may become necessary.
Dialysis removes waste products from the bloodstream when the kidneys can no longer perform this function effectively. In severe cases, a kidney transplant may provide the most effective long term treatment option.
Living with Polycystic Kidney Disease
Living with PKD requires ongoing medical monitoring and proactive health management. Regular checkups allow physicians to assess kidney function, detect complications early, and adjust treatment plans accordingly.
Patients are often encouraged to work closely with nephrologists, follow prescribed medications, and adopt healthy lifestyle practices. Support groups and patient education programs can also help individuals cope with the emotional and physical challenges associated with chronic kidney disease.
Conclusion
Polycystic Kidney Disease is a complex genetic disorder that progressively affects kidney structure and function. Caused primarily by mutations in the PKD1 and PKD2 genes, the disease leads to the formation of multiple cysts that gradually impair the kidneys’ ability to filter waste from the body.
Although PKD cannot currently be cured, early diagnosis and appropriate medical management can significantly slow disease progression and reduce complications. Advances in genetic research, improved diagnostic tools, and emerging therapeutic options continue to enhance the prospects for individuals affected by this condition.
A comprehensive approach that combines medical treatment, lifestyle modifications, and regular monitoring remains essential for preserving kidney health and maintaining quality of life.